




















杨浩(销售总监)
展会咨询QQ: 515616785
手机: 18964878976

01
近日,据印度医疗器械工业协会(AIM)公布的数据显示,2021年,印度医疗器械进口额从2020年的447.08亿卢比(约37.9亿元人民币)增长了41%,达到创纪录的632亿卢比(约53.6亿元人民币)。AIM一位工作人员表示:“这是一个令人担忧的情况,因为在六年的时间里,这一数字增长了五倍。”他说,2016年,印度进口了价值128.66亿卢比(约10.9亿元人民币)的医疗器械。
AIM的数据显示,中国仍是印度医疗器械的主要来源地,与2020年相比,2021年来自中国的医疗器械发货量增长了48%,达到135.38亿卢比(约11.48亿元人民币)。印度医疗器械市场估计超过877.52亿卢比(约77.4亿元人民币),其中海外供应商占70%以上。有专家表示,印度对进口医疗器械的依赖引发了许多担忧,许多医院寻求最新的医疗器械,这部分往往只能依赖进口。

02 03
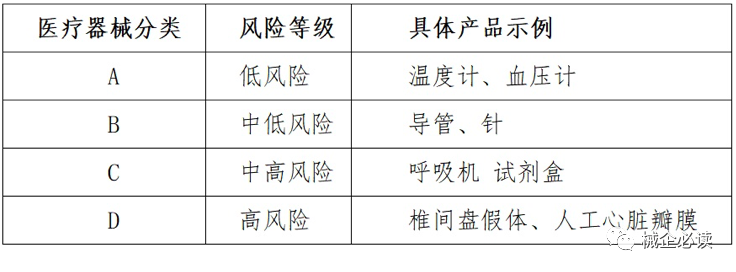
(一)医疗器械的分类
2017年印度对医疗器械法规进行了大幅修订,公布了《医疗器械管理条例2017》(medical device rules 2017),从2018年1月起实施,参照了“全球医疗器械法规调和会”(the global harmonization task force,简称ghtf),将医疗器械分为A到D四类,对应从低风险到高风险四种风险级别。
(二)医疗器械上市路径
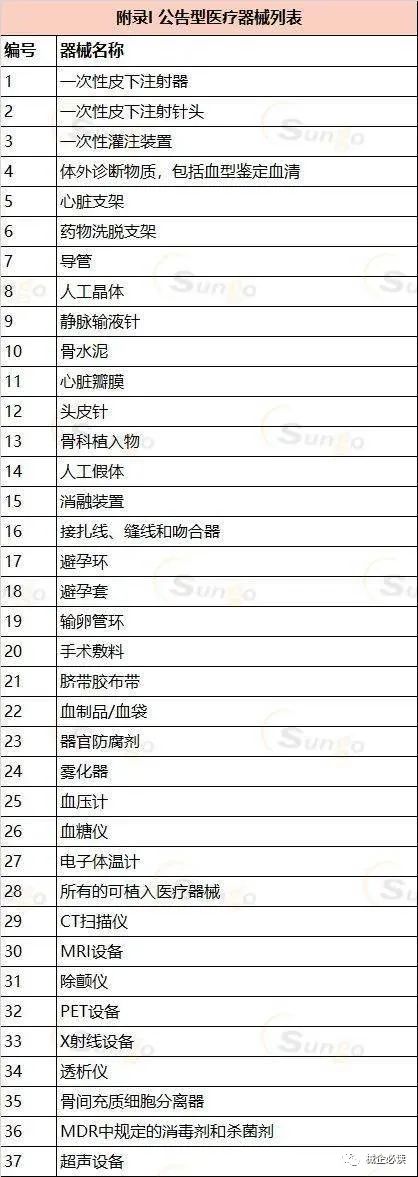
MDR2017及其修正案将目前的医疗器械上市路径分为两种:公告型和非公告型。
公告型医疗器械:法规列出了37个具体的种类,强制要求制造商和进口商分别申请生产许可证和进口许可证。
根据产品的分类,印度主管当局可能会在许可证颁发前后进行生产场地的审核。
(四)进口许可申请
出口医疗器械至印度进行销售或使用,必须经印度当地授权代理人(authorised agent)向cdsco提出医疗器械进口许可证申请(包括a至d级)。cdsco对医疗器械进口许可证的审核主要分为两步:
第一步:确认生产企业质量管理体系(quality management system,简称qms)的符合性
首先cdsco审核授权代理人所提交的生产企业质量管理体系,其必须符合印度医疗器械质量管理标准icmed 13485(修改采用自iso 13485,我国标准yy/t 0287等效采用了iso 13485),包括:生产企业qms技术文件、检测报告、最近一次现场检查报告等,如果cdsco认定生产企业qms的符合存在疑虑时,可以要求实施评估、产品检测或生产企业现场检查(费用由代理人承担)。
第二步:审核医疗器械的安全性与有效性
如果出口印度的医疗器械分类属于a级或者b级,授权代理人应向cdsco提交中国的自由销售证明,或者中国实施临床测试的数据(或其他可供证明产品安全性与有效性的数据)。如果出口印度的医疗器械分类属于c级或者d级,授权代理人必须在印度实施临床测试。
例外情况:如果医疗器械(A级至D级)已由欧盟、美国、加拿大、日本或澳大利亚的监管机构颁发了自由销售证明,则无需再进行临床测试。
2026版权所有:上海展亚展览服务有限公司 医博会、医疗器械展会、医疗展会、北京医疗器械展览会 北京医疗展、医疗器械博览会、医疗博览会、国际医疗器械博览会、CMEH
